Informatics for Material Morphologies using Reinforcement Learning and Active Learning
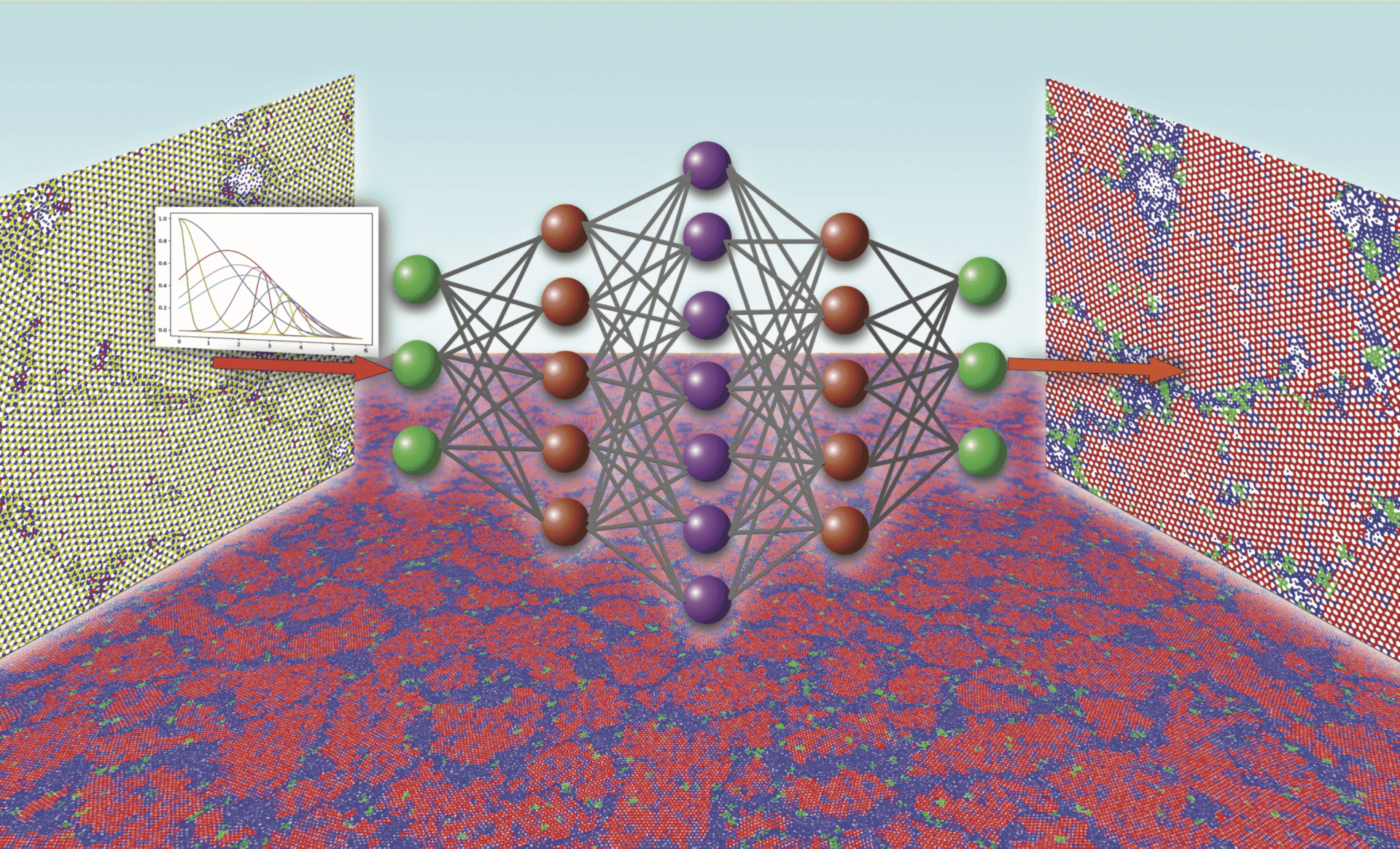 MAGICS develops various artificial intelligence (AI) and machine learning (ML) software for materials, including reinforcement learning (RL) and active learning (AL) to optimize nano-architectures and deep generative models to find transition pathways.
MAGICS develops various artificial intelligence (AI) and machine learning (ML) software for materials, including reinforcement learning (RL) and active learning (AL) to optimize nano-architectures and deep generative models to find transition pathways.
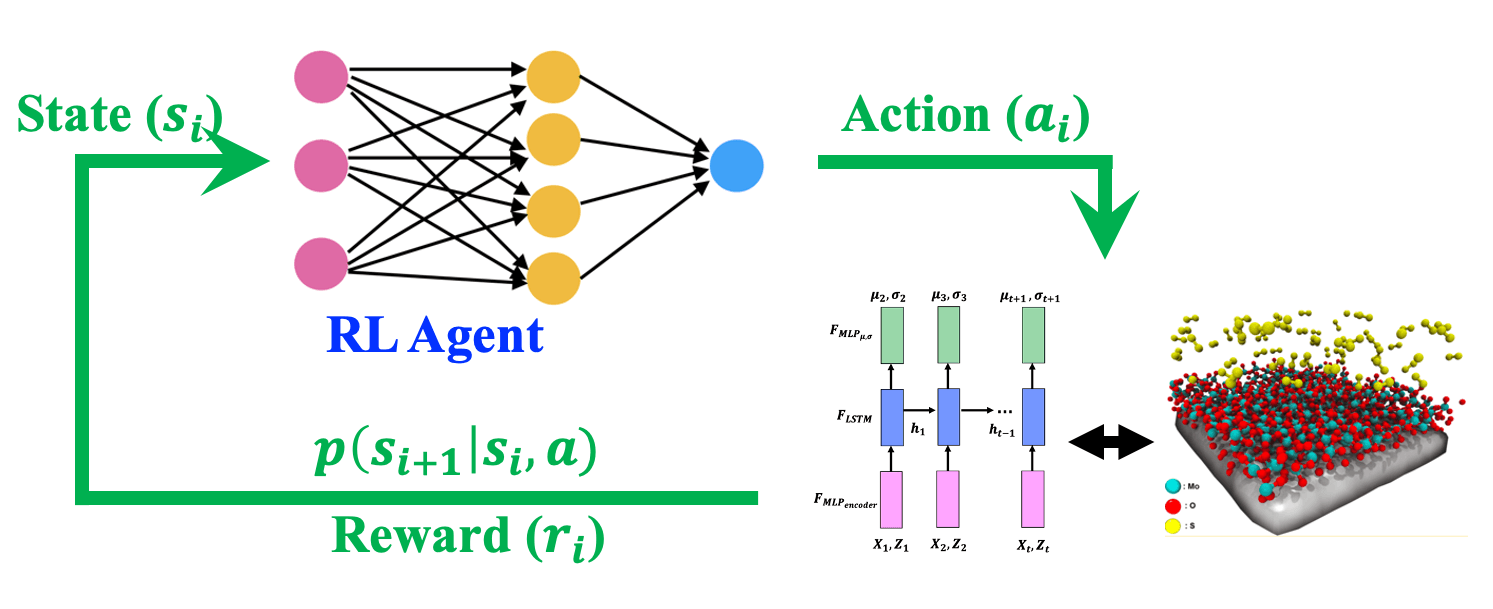
Designing experimental synthesis condition of materials is a challenging task as it involves decision making process of time-dependent synthesis parameters. Further, these experiments take very long time to perform, and thus its not feasible to use the active learning approaches to directly learn the optimal synthesis condition. Here, we have used Offline Model based Reinforcement Leaning (RL) to propose the optimal synthesis condition of 2D quantum material. After trainng, RL agent not only outperformed the random baseline model by more then 50% by desiging optimal policy of synthesis condtiom of reaction temperature and reactant gas concentration but also provided mechanistic insight of synthesis process itself in terms of synthesis parameters
Quantum material synthesis by reinforcement learning, P. Rajak, A. Krishnamoorthy, R. K. Kalia, A. Nakano, and P. Vashishta, Proc. NeurIPS Workshop on Machine Learning and the Physical Sciences, 170 (2020)
Software Repository: TBA
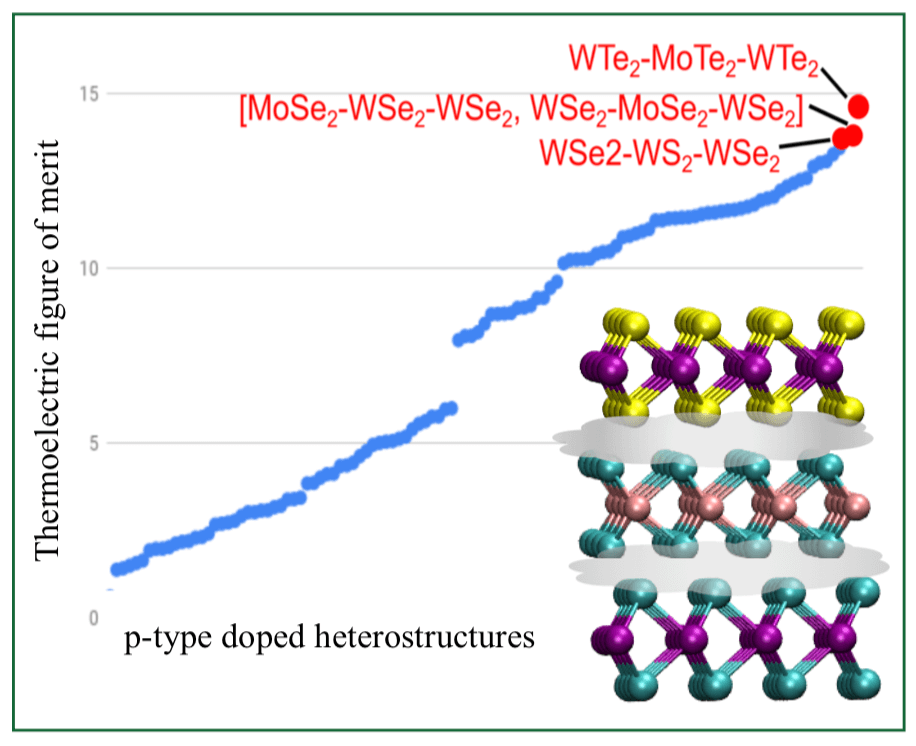 Properies such as band gap and thermoelectric behaviour of vertically stacked 2D materials can be tunned by careully choosing the compostion of materials at each layer. However, the state space of material goes exponentially with the number of layers and qunatim mechanical (QM) calculation of each structure is very expensive and time consuming. Thus, brute force or regression based models are not suitable choice to find the optimial structure from this exponentially large search space. Here, we have developed an active learning model based on Bayesian optimization to find the best stacked 2D material structure with desired properties with minimum QM calculation.
Properies such as band gap and thermoelectric behaviour of vertically stacked 2D materials can be tunned by careully choosing the compostion of materials at each layer. However, the state space of material goes exponentially with the number of layers and qunatim mechanical (QM) calculation of each structure is very expensive and time consuming. Thus, brute force or regression based models are not suitable choice to find the optimial structure from this exponentially large search space. Here, we have developed an active learning model based on Bayesian optimization to find the best stacked 2D material structure with desired properties with minimum QM calculation.
Software Repository: Github
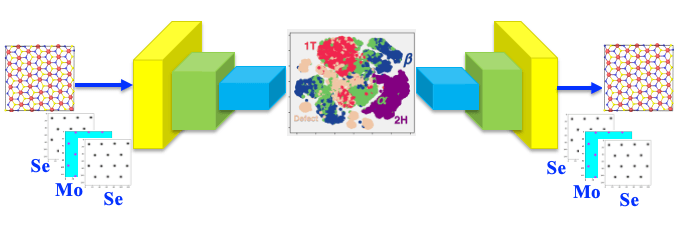 Learning the mechanism of phase transformation in matwrials is a complex and time consuming process. Here, we have developed a generative model of the phase transformation pathways in MoWSe2 during dyanamic fracture using variational autoencoder (VAE). The trained VAE model correctly identifies transformation pathways connecting the semiconducting (2H) and metallic (1T) phases via novel intermediate structures, which is also observed experimently. Further, the mechanistic insight of transformation provided by VAE is much faster then tradtional molecular dynamics simulation that takes 2-3 days.
Learning the mechanism of phase transformation in matwrials is a complex and time consuming process. Here, we have developed a generative model of the phase transformation pathways in MoWSe2 during dyanamic fracture using variational autoencoder (VAE). The trained VAE model correctly identifies transformation pathways connecting the semiconducting (2H) and metallic (1T) phases via novel intermediate structures, which is also observed experimently. Further, the mechanistic insight of transformation provided by VAE is much faster then tradtional molecular dynamics simulation that takes 2-3 days.
Structural phase transitions in MoWSe2 monolayer - molecular dynamics simulations and variational autoencoder analysis, P. Rajak, A. Krishnamoorthy, A. Nakano, P. Vashishta, and R. K. Kalia, Physical Review B 100, 014108: 1-7 (2019)
Software Repository: Github
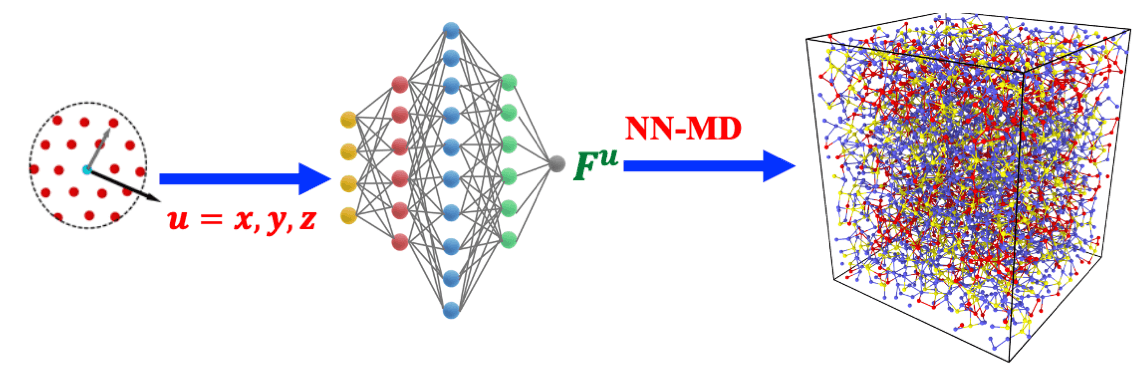
Neural network molecular dynamics (NNMD) simulations could revolutionize atomistic modeling of materials with quantum-mechanical accuracy at a fraction of computational cost. We have developed a scalable parallel NNMD software (RXMD-NN) based on RXMD. RXMD-NN has achieved high scalability up to 786,432 IBM BlueGene/Q cores involving 1.7 billion atoms. Furthermore, we have achieved 4.6-fold reduction of T2S by using a novel network that directly predicts atomic forces from feature vectors.
Neural network molecular dynamics at scale, P. Rajak, K. Liu, A. Krishnamoorthy, R. K. Kalia, A. Nakano, K. Nomura, S. C. Tiwari, and P. Vashishta, Proc. IPDPS Workshop on Scalable Deep Learning over Parallel and Distributed Infrastructure, ScaDL (2020) p. 991
Software Repository: Github