RXMD
RXMD is a scalable parallel software for reactive molecular dynamics (RMD) simulations based on the first principles-informed reactive force-field (ReaxFF) approach. RMD follows the time evolution of atomic trajectories, where ReaxFF describes chemical bond breakage and formation based on a reactive bond-order concept and charge transfer between atoms based on a charge-equilibration approach.
RXMD Software Repository
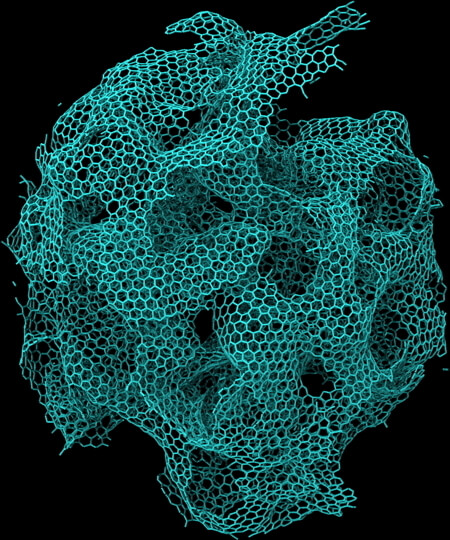
Computational synthesis of fractal nanocarbon by oxidation of a silicon-carbide nanoparticle. 112 million-atom RMD simulation was performed on 786,432 IBM Blue Gene/Q cores.
References
- A scalable parallel algorithm for large-scale reactive force-field molecular dynamics simulations, K. Nomura, R. K. Kalia, A. Nakano, and P. Vashishta, Computer Physics Communications 178, 73 (2008).
- An extended-Lagrangian scheme for charge equilibration in reactive molecular dynamics simulations, K. Nomura, P. E. Small, R. K. Kalia, A. Nakano, and P. Vashishta, Computer Physics Communications 192, 91 (2015).
- Nanocarbon synthesis by high-temperature oxidation of nanoparticles, K. Nomura, R. K. Kalia, Y. Li, A. Nakano, P. Rajak, C. Sheng, K. Shimamura, F. Shimojo, and P. Vashishta, Scientific Reports 6, 24109 (2016).